In this work, the enantioselective total synthesis of (–)-quinine has been accomplished in a pot-economical manner using five reaction vessels. In the first pot, reactions involve the diphenylprolinol silyl ether-mediated Michael reaction, aza-Henry reaction, hemiaminalization, and elimination of HNO2 (five reactions), affording a chiral tetrahydropyridine with excellent enantioselectivity. In the second pot, five reactions proceed with excellent diastereoselectivity to afford a trisubstituted piperidine with the desired stereochemistry. A further five reactions are carried out in the last one-pot sequence.

Quinine is one of the cinchona alkaloids; it is extracted from the bark of cinchona trees 1 . It is an important antimalarial medication 2 . Its derivatives are used not only as ligands of asymmetric metal catalysts, but also as effective asymmetric organocatalysts 3,4,5,6,7,8,9,10 . Quinine has fascinated synthetic organic chemists for several decades 11,12,13,14,15,16,17,18,19,20,21,22,23,24,25,26,27,28 . It contains four chiral centers, and stereoselective synthesis is a challenge.
In 1944, Woodward and Doering synthesized homomeroquinene—the first formal synthesis of a racemic quinine 4,5 . Stork then reported the first synthesis of chiral (–)-quinine from a chiral starting material using highly diastereoselective reactions 17 . Jacobsen 18 and Kobayashi 20 independently synthesized (–)-quinine by constructing a quinuclidine skeleton, by a C8–N bond formation, using an epoxide as an electrophile. Jacobsen used an asymmetric catalytic reaction for the enantioselective synthesis. Aggarwal used a chiral Corey–Chaykovsky reaction as a key step 22,23 , while Maulide used a C–H activation reaction and an aldol reaction 25 . Chen synthesized quinine and quinidine using a local desymmetrization-based strategy 26 .
Recently, organocatalysts have been used for the synthesis of quinine. Hatakeyama used a proline-mediated intramolecular aldol reaction for the formation of a piperidine skeleton 24 , while the Michael reaction of dimethyl malonate and α,β-unsaturated aldehyde catalyzed by diphenylprolinol silyl ether is a key step in Córdova’s synthesis 27 . Ishikawa has reported a synthesis of (–)-quinine involving a one-pot synthesis of a chiral piperidine skeleton by a diphenylprolinol silyl ether-mediated formal aza-[3 + 3] cycloaddition/Strecker-type cyanation reaction 28 . Although there are several asymmetric total syntheses of (–)-quinine, it is still desirable to develop an efficient and practical method for its synthesis. Moreover, a method that facilitates derivatization is desirable, particularly because of its importance as a medication and as an organocatalyst.
One-pot operations are effective methods for making several bonds and generating a complexity of molecules in a single-pot sequence 29,30,31 . Moreover, one-pot operations circumvent several purification steps via in situ quenching events, thereby minimizing chemical waste generation and saving time. Based on this, we proposed the concept of “pot economy.” We subsequently accomplished several total syntheses based on this concept 32,33,34,35,36,37,38 .
In this work, we describe the “pot-economical” enantioselective total synthesis of (–)-quinine using organocatalyst-mediated reaction.
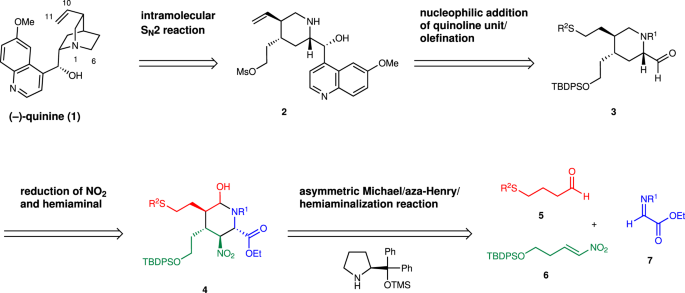
The retrosynthetic analysis of (–)-quinine is shown in Fig. 1. It would be synthesized by making a C6–N bond from a piperidine precursor 2 via an intramolecular SN2 reaction. 2 would be prepared by the addition reaction of a quinoline unit to piperdinecarbaldehyde 3. 3 would be synthesized from a substituted piperidine derivative 4 by functional group transformation. 4 would be prepared by a three-component coupling reaction of aldehyde 5, nitroalkene 6, and imine 7, as developed by our group 39 .
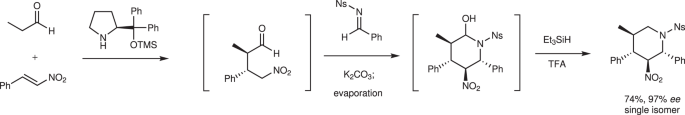
Earlier, we reported a one-pot synthesis of a piperidine skeleton via a sequential reaction of the diphenylprolinol silyl ether-mediated Michael reaction 40 , aza-Henry reaction, and hemiaminalization reaction (Fig. 2). As simple starting materials were used in the earlier study, we considered it now a challenge to determine whether the key reaction proceeds using densely functionalized substrates and whether high stereoselectivity is realized.
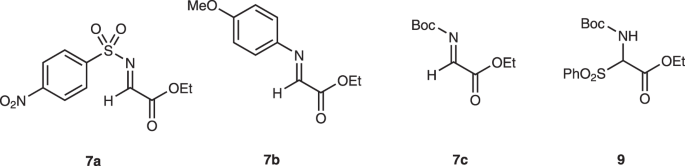
4-Arylthiobutanal 5 41 was selected as a starting aldehyde, taking into consideration of the required introduction of a C10–C11 double bond in a latter stage of the synthesis. The asymmetric Michael reaction of 5 and nitroalkene 6, catalyzed by diphenylprolinol silyl ether, proceeded to afford 8 in good yield and with excellent diastereo- and enantioselectivities (Fig. 3). However, the next aza-Henry reaction was found to be problematic. In our previous study, we used p-nitrophenylsulfonyl (Ns) 42 imine generated from arylaldehyde (Fig. 2). The Ns imine of ethyl glyoxylate 7a is too unstable and difficult to handle (Fig. 4). Imine 7b derived from p-anisidine is too stable to react with 8. On the other hand, the corresponding N-Boc imine 7c possesses suitable stability and reactivity, but it is difficult to isolate it. Thus, it was generated in situ from the corresponding sulfonyl precursor 9 43 with DBU. A domino reaction of 8 and 9, involving the generation of the imine and the aza-Henry reaction, proceeded well to afford piperidine 4 and tetrahydropyridine 10 in 35% and 50% yield, respectively (Fig. 5). Although 4 was an expected product, 10 would be a more suitable intermediate for the total synthesis, because of the harsh reaction conditions that are usually necessary for the reductive removal of a NO2 group from 4 44,45,46 . 4 can be converted to 10 by the treatment with DBU.
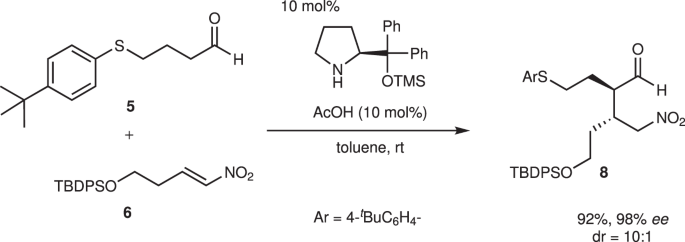
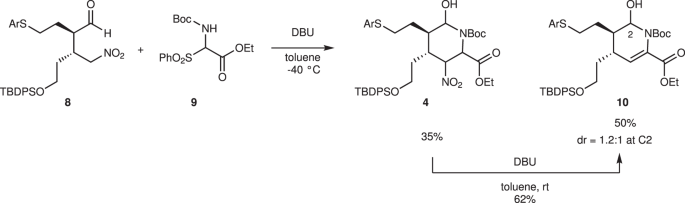
These reactions (5 + 6→10) can be conducted in a single pot (Fig. 6). The first pot reaction starts from the Michael reaction. The asymmetric Michael reaction of 5 and 6 proceeded to afford product 8. Upon the addition of imine precursor 9 and DBU to the same vessel, a domino reaction proceeded. That is, in situ imine generation occurred, and an aza-Henry reaction proceeded, sequentially, followed by hemiaminalization to afford 4. Elimination of HNO2 with DBU afforded 10 with a mixture of diastereomers at C2. Good overall yield, high diastereoselectivity (dr = 10:1 at C3), and excellent enantioselectivity (98% ee) were obtained. The reaction proceeded deca-gram scale efficiently. It should be noted that DBU plays three roles as a base in this transformation: (1) to generate imine (9→7c), (2) to promote the aza-Henry reaction, and (3) to eliminate HNO2.
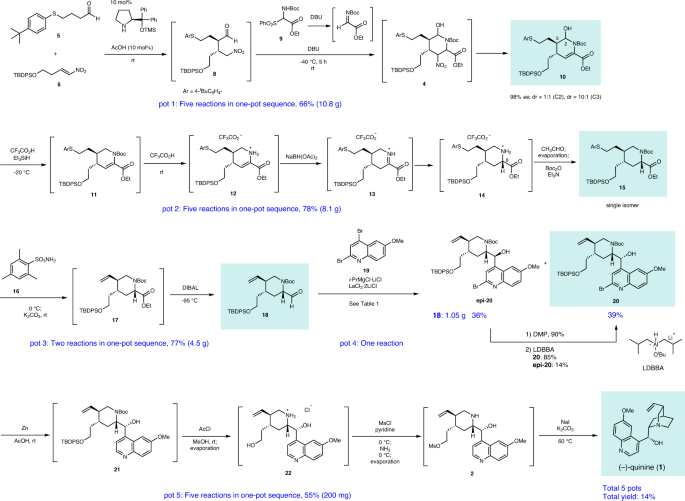
The second pot reaction starts with the reductive removal of a hydroxy group at C2 by the treatment of 10 with Et3SiH and CF3CO2H (TFA) at –20 °C 39 . Upon increasing the temperature of a reaction mixture to room temperature, the deprotection of a Boc group took place, to afford an enamine 12, which was tautomerized to afford an iminium ion 13. Upon the addition of NaBH(OAc)3 to the reaction vessel, stereoselective reduction occurred to afford 14 17 . Addition of CH3CHO led to decomposition of the remaining reducing reagent, and the excess CH3CHO and TFA were removed under reduced pressure. Thus, through this in situ quenching method 30 , a one-pot sequential reaction can be realized.
Further addition of Boc2O and Et3N to the reaction mixture afforded a highly substituted piperidine derivative 15 as a single isomer with the desired configuration. It should be noted that TFA plays three roles as an acid. It facilitates the formation of tetrahydropyridium from hemiaminal 10 at low temperature (–20 °C), to deprotect the Boc group at higher temperature, and to generate the iminium ion 13. It is synthetically efficient that the same reagent plays several roles in a one-pot reaction 29 . The reaction proceeded in a gram scale and the overall yield of this second pot (10→15) was 78%. In this transformation, procedures of deprotection and reprotection of Boc group were involved. The stereoselective reduction of Boc protected alkene 11 did not proceed well, while stereoselective reduction of the iminium ion 13 was successful. Thus, the deprotection step of Boc group is necessary.
We employed two reducing reagents such as Et3SiH and NaBH(OAc)3 for the reduction of hemiaminal (10→11) and iminium ion (13→14). It would be synthetically simple to use a single reducing reagent in a one-pot reaction. Although Et3SiH can act as a reducing reagent in both reactions, the second reaction was slow in the low yield with the partial deprotection of TBDPS group. In the case of NaBH(OAc)3 the reaction from 10 to 11 did not proceed well with the generation of several byproducts. On the other hand, the sequential use of two reducing reagents gave a good result in the second pot reaction.
The third pot involves the preparation of a C10–C11 alkene and half-reduction of an ester. When arylthiol ether 15 was treated with 16, according to Matsuo’s protocol, alkene 17 was generated 47 . In the same reaction vessel, the addition of DIBAL at low temperature (–95 °C) reduced an ester, and aldehyde 18 was obtained in good yield (77%) over two steps, in one pot. The conventional method of arylsulfanylethyl moiety into vinyl substituent is a two-step process: oxidation to sulfoxide and elimination of ArSOH. However, by applying Matsuo’s protocol, it becomes a one-step process. Moreover, the reaction conditions here are comparable to those of the one-pot sequential reaction of the next DIBAL reduction. This step is also scalable.
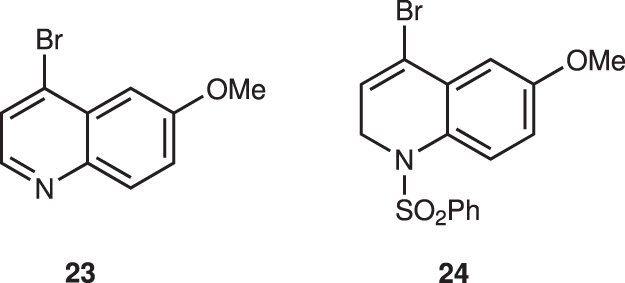
The next step is the introduction of a quinoline moiety to aldehyde 18—this was found to be very difficult. According to Ishikawa, several quinoline metals generated from 23 did not react with a similar aldehyde with formation of dimer of 23 because the C2 position of quinoline is electrophilic—thus, dihydroquinoline lithium reagent generated from 24 was developed as a nucleophile (Fig. 7) 28 . In order to synthesize C2’ derivatives of quinine and also to reduce the electrophilicity at the C2 of quinoline, we used 2,4-dibromo-6-methoxyquinoline (19) as a quinoline precursor. However, the selective halogen–metal exchange reaction of Br at C4 over Br at C2 is another concern. Pleasingly, the selective halogen–metal exchange of Br at C4 occurred selectively. But neither lithium nor magnesium species were added to aldehyde 18 (Table 1; entries 1, 2).
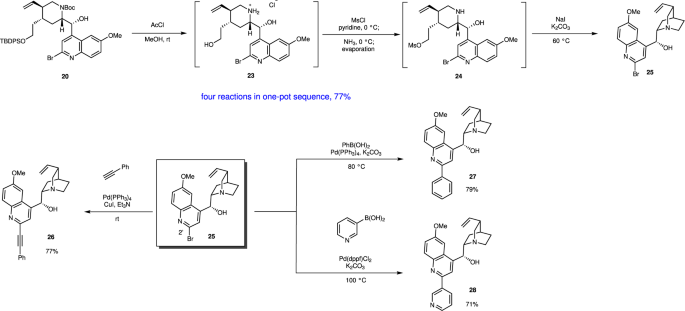
In summary, an efficient, enantioselective, and pot-economical total synthesis of (–)-quinine has been accomplished. The present synthesis has several noteworthy features. (1) A highly functionalized chiral tetrahydropyridine can be synthesized in the first one-pot operation, which consists of a succession of five reaction steps, including a diphenylprolinol silyl ether-mediated asymmetric Michael reaction as developed in our group, a domino aza-Henry reaction/hemiacetal formation involving the in situ generation of imine, and elimination of HNO2. (2) Five reactions can be successfully conducted in the second one-pot operation: the reductive removal of a hydroxy group, deprotection of Boc, iminium ion generation, stereoselective reduction, and Boc protection, thus affording the trisubstituted piperidine as a single isomer with the correct configuration. (3) Five reactions can proceed efficiently by careful choice of the reaction order and the reaction conditions: the reductive removal of Br, deprotection of Boc and TBDPS groups, selective synthesis of a mono-Ms derivative, and an intramolecular SN2 reaction. (4) The use of 2,4-dibromo-6-methoxy quinoline for the introduction of a quinoline moiety makes feasible the derivatization at the C2′ position of (–)-quinine.
This synthesis requires a total of five separate one-pot operations and five purifications by column chromatography, plus two additional operations for the conversion of the undesired epi-20 to the desired 20. The total yield of (–)-quinine was 14%.
All reactions were carried out under argon atmosphere and monitored by thin-layer chromatography using Merck 60 F254 precoated silica gel plates (0.25 mm thickness). Specific optical rotations were measured using a JASCO P-2200 polarimeter. FT-IR spectra were recorded on a JASCO FT/IR-4600HC1 spectrometer. 1 H and 13 C NMR spectra were recorded on an Agilent-400 MR (400 MHz for 1 H NMR, 100 M Hz for 13 C NMR) instrument. Data for 1 H NMR are reported as chemical shift (δ ppm), integration multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, septet = sep, dd = doublet of doublets, ddd = doublet of doublet of doublets, ddt = doublet of doublet of triplets, dt = doublet of triplets, dq = doublet of quartets, m = multiplet, brs = broad singlet, brd = broad doublet, brt = broad triplet), coupling constant (Hz), Data for 13 C NMR are reported as chemical shift. High resolution ESI-TOF mass spectra were measured by Themo Orbi-trap LTQ XL instrument. HPLC analysis was performed on a HITACHI Elite LaChrom Series HPLC, UV detection monitored at appropriate wavelength respectively, using CHIRALPACK® IB (0.46 cm × 25 cm). Melting point was measured using Yanaco MP-J3. Flash chromatography was performed using silica gel 60 N (spherical) or silica gel 60 N (spherical) NH2 of Kanto Chemical Co. Int., Tokyo, Japan. All reagents were purchased from commercial sources (Aldrich, FUJIFILM Wako chemicals, Kanto Chemical, TCI).
Experimental procedures, characterizations, spectra are available in the Supplementary Information.
This work was supported by JSPS KAKENHI Grant Number JP19H05630 (Y.H.).